|
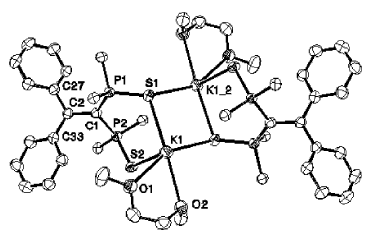
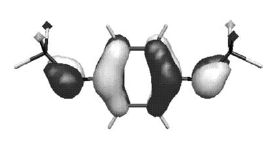
Chemical and electrochemical reductions of the macrocycle 1 lead to the formation of a radical monoanion anion [1]â¢- whose structure has been studied by EPR in liquid and frozen solutions. In accord with experimental 31P hyperfine tensors, DFT calculations indicate that, in this species, the unpaired electron is mainly localized in a bonding Ï PâP orbital. Clearly, a one-electron bond (2.763 Ã
) was formed between two phosphorus atoms which, in the neutral molecule, were 3.256 Ã
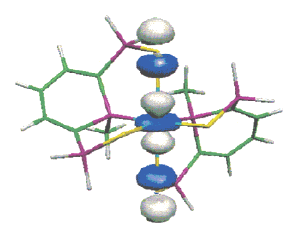
apart (crystal structure). A subsequent reduction of this radical anion gives rise to the dianion [1]2-Â which could be crystallized by using, in the presence of cryptand, Na naphthalenide as a reductant agent. As shown by the crystal structure, in [1]2-, the two phosphinine moieties adopt a phosphacyclohexadienyl structure and are linked by a PâP bond whose length (2.305(2) Ã
) is only slightly longer than a usual PâP bond. When the phosphinine moieties are not incorporated in a macrocycle, no formation of any one-electron PâP bond is observed: thus, one-electron reduction of 3 with Na naphthalenide leads to the EPR spectrum of the ion pair [3]â¢- Na+; however, at high concentration, these ion pairs dimerize, and, as shown by the crystal structure of [(3)2]2-[{Na(THF)2}2]2+ a PâP bond is formed (2.286(2) Ã
) between two phosphinine rings which adopt a boat-type conformation, the whole edifice being stabilized by two carbonâsodiumâphosphorus bridges. |